DNA-Metabarcoding
Klassischer Weise erfolgt die Artbestimmung von Insekten auf morphologischer Basis: Sie werden durch Spezialisten lebend im Freiland bestimmt oder aber gefangen und präpariert; die Bestimmung erfolgt dann im Labor unter zu Hilfenahme von Mikroskop, Bestimmungsliteratur und bereits sicher bestimmten Vergleichsexemplaren. Für sehr viele Insektengruppen ist diese Methode jedoch praktisch undurchführbar: es gibt entweder nicht genügend Experten oder der finanzielle Aufwand wäre zu hoch.
Die noch junge Methode der genetischen Artbestimmung mittels DNA-Metabarcoding stellt eine elegante Lösung dieses Dilemmas dar. Sie befindet sich aber noch im Aufbau, was bei der Interpretation der Daten zu berücksichtigen ist. In Einzelfällen kann es beim jetzigen Stand der Methode noch zu Fehlbestimmungen kommen. Sollte also beispielsweise in einer gut bearbeiteten Gruppe eine Art weitab ihres üblichen Verbreitungsgebietes nachgewiesen werden, dann sollte der Nachweis im Zweifel geprüft werden. Andererseits handelt es sich bei den meisten „Erstnachweisen“ in schlecht bearbeiteten Gruppen mit Sicherheit um korrekte Daten – so wird beispielsweise geschätzt, dass es sich bei den bislang aus Deutschland bekannten ca. 1600 Erzwespen-Arten (Chalcidoidea) nur um die Hälfte der tatsächlich vorkommenden Arten handelt und dass allein in der Familie der Gallmücken (Cecidomyiidae) noch mehrere tausend Arten zu entdecken sind.
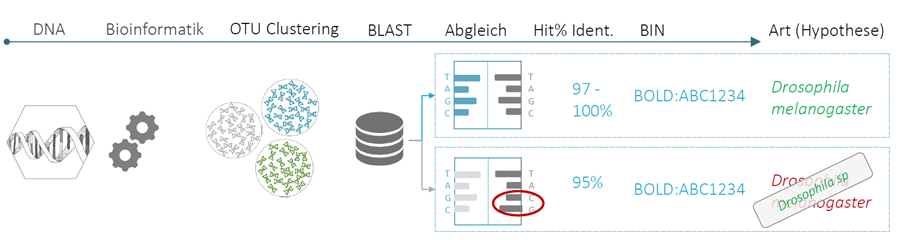
In vereinfachter Form dargestellt beinhaltet das Metabarcoding die folgenden Arbeitsschritte: Das in Ethanol konservierte Tiermaterial wird getrocknet, pulverisiert und anschließend wird aus einer Teilprobe die DNA extrahiert (erster Schritt „DNA“ in der unteren Abbildung). Im Rahmen eines spezifischen bioinformatorischen Prozesses („Bioinformatik“) werden aus den gewonnenen DNA-Sequenzen zunächst sogenannte OTUs (Operational Taxonomic Units) gebildet („OTU Clustering“). Diese wiederum werden in einem zweiten Schritt („BLAST“) mit den bereits bekannten und in einer weltweiten Datenbank (dem Barcode of Life Data System, kurz BOLD) erfassten Barcodes abgeglichen („Abgleich“). Stimmen die Sequenzen zu mindestens 97 % überein („Hit%Ident“), kann davon ausgegangen werden, dass die verglichenen Sequenzen zur selben Art gehören. Bei geringeren Übereinstimmungen ist dies nicht der Fall und man erhält je nach Übereinstimmungsgrad nur die Information, zu welcher Gattung (95-97 %), Familie (90-95 %), Ordnung (85-90 %) oder Klasse (80-85 %) die Sequenz gehört. Als Artnachweise werden generell nur Sequenzen mit mindesten 97 % Übereinstimmung berücksichtigt.
BOLD nutzt zur Identifikation von DNA-Sequenzen bzw. Barcodes, unabhängig von ihrer taxonomischen Zuordnung, sogenannte BINs (Barcode Index Nummern). BINs basieren auf einer Kette von Algorithmen, die ähnliche DNA-Sequenzen gruppieren und dabei auf taxonomische Integrität hin überprüfen. In 98 % aller Fälle entsprechen die so berechneten BINs konkreten Arten. Somit können BINs stellvertretend als Arten angesehen werden („BIN-Arten“), unabhängig davon, ob ihnen schon ein wissenschaftlicher Artname zugeordnet wurde oder nicht. Es kann vorkommen, dass sich nah verwandte Arten nicht trennen lassen, sondern der gleichen BIN zugeordnet werden („BIN sharing“ aufgrund geringer interpezifischer Variation der DNA-Sequenzen). In diesen Fällen liefert die Methode bislang nur die Information, zu welcher Gattung die Art gehört, nicht aber um welche Art es sich genau handelt. Ebenso kommt es vor, dass einer Art mehrere BINs zugeordnet werden („multiple BINs“ aufgrund hoher intraspezifischer Variation der DNA-Sequenz). In diesen Fällen ist es möglich, dass es sich in Wirklichkeit um mehrere Arten handelt („cryptic species“).
In welchem Umfang die gesammelten DNA-Sequenzen konkreten Arten oder zumindest BIN-Arten zugeordnet werden können, hängt in erster Linie davon ab, ob die entsprechenden Arten bzw. ihre Barcodes schon in BOLD enthalten sind oder nicht. Der diesbezügliche Erfassungsgrad unterscheidet sich zur Zeit noch stark zwischen den verschiedenen Insektengruppen. Während beispielsweise die mitteleuropäischen Schmetterlinge (Lepidoptera) bereits beinahe vollständig erfasst wurden, ist dies bei den parasitoiden Wespen noch lange nicht der Fall: Diese gehören zu den taxonomisch schwierigsten und zugleich artenreichsten Insektengruppen überhaupt, sie sind bekanntermaßen selbst in Mitteleuropa noch nicht vollständig erforscht. An diesem Punkt kommt jedoch ein wesentlicher Vorteil des DNA-Barcodings zum tragen: die gewonnenen Sequenzen bzw. Barcodes können in Zukunft immer wieder neu abgeglichen werden. Da BOLD praktisch mit jedem Tag größer und vollständiger wird, wird eine zukünftige erneute Abfrage der Daten ein detaillierteres und genaueres Bild ergeben als zum jetzigen Zeitpunkt. Die jetzt gewonnenen Barcoding-Daten können also als Basis für ein zukünftiges Biodiversitätsmonitoring genutzt werden, das in Zeiten des Insektensterbens dringend benötig wird.
Durchgeführt wird das DNA-Metabarcoding durch die Firma AIM – Advanced Identification Methods.

Für die morphologische Artbestimmung wird in der Regel eine Vergleichssammlung mit sicher bestimmten Exemplaren benötigt.

Für das DNA-Metabarcoding werden die Insekten zunächst in Alkohol konserviert, später aber in der Regel vollständig zerstört, so dass keine Belegexemplare zurückbleiben.
Das DNA-Metabarcoding ist zwar ein im Detail sehr aufwändiger Prozess – im Vergleich zur morphologischen Artbestimmung können aber sehr große Insektenmengen mit vergleichsweise sehr geringem Aufwand bearbeitet werden.